Importing count data from Primary Analysis
If you have run the Primary analysis workflow and would like to import the counts file from the Primary analysis workflow and use it to conduct differential expression analysis and pathway analysis, use the steps below -
Note
From your Primary analysis history, you will notice one of the results collection from featureCounts named as “featureCounts on collection N: Counts”
This is the folder containing the counts for each of your samples. If you used the example dataset, the files should appear as GSM461176_untreat, GSM461179_treat, and GSM461182_untreat
Scroll to the top of the history, and select the settings button
Under Dataset options, Click on the option “Copy Datasets”
This will open a new page where you can check the box next to the dataset you want to copy to a new history. Refer to the screenshot 1 and 2 below.
Select the “featureCounts on collection N: Counts” folder and select the “Destination history” as the history you created earlier - “RNA-seq tertiary analysis”
You will need three more files - “KEGG pathways” to provide pathways to goseq, “header” file to create a header file to attach to DESeq2, and the Drosophila gene annotation file to run this workflow.
For your organism, download the GTF file using the steps from “Primary Analysis” into this history. For the Drosophila gene annotation file, download the file “Drosophila_melanogaster.BDGP6.87.gtf” and upload it to the history
For the header file, download the file (Above “History”, click on the three dots and download the file) from this page and upload the file to Galaxy using instructions from here
For the KEGG pathway file, download the file from this page and upload the file to Galaxy using instructions from here
You can now ready to run the Tertiary analysis workflow and go through the next pages to improve your understanding of each step
Go to workflows > “RNA-seq Tertiary Analysis” and click on the run button
Under “Input Dataset Collection”, select your collection of counts files
Under GTF file - gene annotation, select your gene annotation file from your history
Under “KEGG pathways to plot”, select the KEGG pathway file from your history
Under “header”, select the header file from your history
Another important note is for the “goseq” tool and “Pathview” tool. Depending on the gene ID format in your GTF file, scroll down to the two goseq tools in the workflow, expand each, and check the format under “Select Gene ID format”. Use the button next to it to select your gene ID format from the dropdown list. Repeat this for the “Pathview” tool as well
Before you run the workflow, please review the changes you have to make to the DESeq2 tool instructions in order to run it with your dataset. Refer to screenshot 3 below if you have treated vs. untreated samples.
Scroll to the top and select “Yes” to send the results to a new history in order to send your workflow results to a new history and rename it so it’s intuitive to you. Screenshot 4 shows how your workflow should look like before you submit.
Click on “Run Workflow”
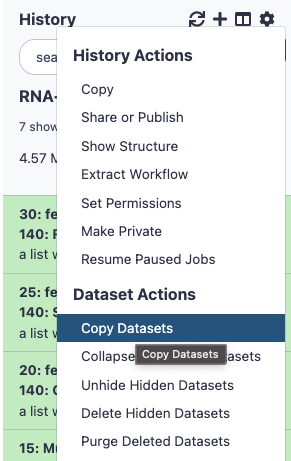
Screenshot 1 - The screenshot shows where to find the “Copy Datasets” option in the history

Screenshot 2 - The screenshot shows how to copy datasets between two histories (copying featureCounts folder from the previous history to the new history)
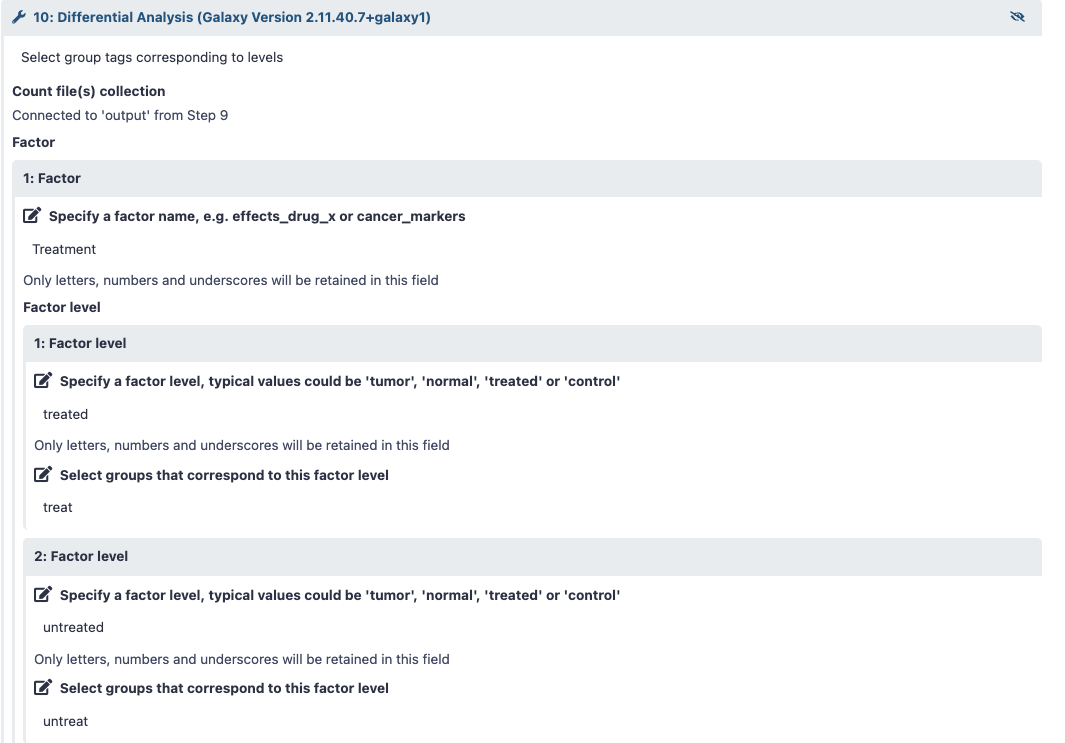
Screenshot 3 - The screenshot shows how to change the DESeq2 options

Screenshot 4 - The screenshot shows how your Tertiary workflow should look with the files selected